XEASY Spin system identification: Difference between revisions
Jump to navigation
Jump to search
No edit summary |
No edit summary |
||
| Line 16: | Line 16: | ||
#In XEASY, use <tt>ns</tt>, <tt>ll</tt> and <tt>ls</tt> to load 2D [15N,1H]-HSQC spectrum, XEASY library and <nop>SequenceList. To change display from the default one to contour plot, type <tt>cp</tt> and give the appropriate threshold level. A corresponding initial <nop>AtomList is generated automatically; use <tt>in</tt> for in-phase 'peak picking' of the 2D [15N,1H]-HSQC spectrum automatically; complete peak picking manually by using <tt>dp</tt> to remove the peaks belonging to sidechain amides which can be identified by a NH2only HSQC if the region is crowded and <tt>pp</tt> to 'peak pick' additional peaks (Figure.1A); Use <tt>ar</tt> to automatically assign each peak to the backbone amide moiety of '''SRD-I''' residues (Figure.1B,C); use <tt>ac</tt>, <tt>wc</tt> and <tt>wp</tt> to save the updated <nop>AtomList as <tt>nhsqcO1.prot</tt> and <nop>PeakList as <tt>nhsqcO1.peaks</tt>. Then, 15N / amide 1HN chemical shifts are transferred into the <nop>AtomList entries corresponding to '''SRD-I'''. At times the <tt>ar</tt> command gives an error "Atom N 201 not known!". This can be rectified by loading a different library file [ll] from /nsm/chem/cen2/HTP2/3_src/xeasy/src.new/xeasy.lib and then trying to auto assign the peaks. <br><br>'''Figure 1.''' Peak picking the NHSQC spectrum* | #In XEASY, use <tt>ns</tt>, <tt>ll</tt> and <tt>ls</tt> to load 2D [15N,1H]-HSQC spectrum, XEASY library and <nop>SequenceList. To change display from the default one to contour plot, type <tt>cp</tt> and give the appropriate threshold level. A corresponding initial <nop>AtomList is generated automatically; use <tt>in</tt> for in-phase 'peak picking' of the 2D [15N,1H]-HSQC spectrum automatically; complete peak picking manually by using <tt>dp</tt> to remove the peaks belonging to sidechain amides which can be identified by a NH2only HSQC if the region is crowded and <tt>pp</tt> to 'peak pick' additional peaks (Figure.1A); Use <tt>ar</tt> to automatically assign each peak to the backbone amide moiety of '''SRD-I''' residues (Figure.1B,C); use <tt>ac</tt>, <tt>wc</tt> and <tt>wp</tt> to save the updated <nop>AtomList as <tt>nhsqcO1.prot</tt> and <nop>PeakList as <tt>nhsqcO1.peaks</tt>. Then, 15N / amide 1HN chemical shifts are transferred into the <nop>AtomList entries corresponding to '''SRD-I'''. At times the <tt>ar</tt> command gives an error "Atom N 201 not known!". This can be rectified by loading a different library file [ll] from /nsm/chem/cen2/HTP2/3_src/xeasy/src.new/xeasy.lib and then trying to auto assign the peaks. <br><br>'''Figure 1.''' Peak picking the NHSQC spectrum* | ||
'''A: After in-phase peak picking; ''' [[ | '''A: After in-phase peak picking; ''' [[Image:XEASY hsqc2.jpg]] <br> | ||
<br> | |||
'''B: XEASY <tt>ar</tt> window''' | '''B: XEASY <tt>ar</tt> window''' | ||
[[ | [[Image:XEASY hsqc ar.jpg]] | ||
<br> | <br> | ||
| Line 33: | Line 32: | ||
<br> | <br> | ||
[[File | [[File:XEASY_hsqc3.jpg]] | ||
#In UBNMR run <tt>makeHncoPeak</tt>. A starting peak list for analysis of (3,2)D HNNCO or 3D HNNCO is generated as <tt>hncoI1.peaks</tt>. / | #In UBNMR run <tt>makeHncoPeak</tt>. A starting peak list for analysis of (3,2)D HNNCO or 3D HNNCO is generated as <tt>hncoI1.peaks</tt>. / | ||
| Line 43: | Line 42: | ||
#In the spectra folder, make a identical copy of HNCO spectrum, eg. cp spectrum <tt>HNCO1</tt> to spectrum <tt>HNCO1a</tt>. | #In the spectra folder, make a identical copy of HNCO spectrum, eg. cp spectrum <tt>HNCO1</tt> to spectrum <tt>HNCO1a</tt>. | ||
#In UBNMR, run <tt>makeHncoPeak</tt> to produce the file <tt>hncoI1.peaks</tt>. This <nop>PeakList will contain one peak for each amide N,H moiety assigned to SRD-I derived from the 2D [15N,1H]-HSQC <nop>PeakList. 175 ppm is assigned to all 13C' shifts of SRD-II. | #In UBNMR, run <tt>makeHncoPeak</tt> to produce the file <tt>hncoI1.peaks</tt>. This <nop>PeakList will contain one peak for each amide N,H moiety assigned to SRD-I derived from the 2D [15N,1H]-HSQC <nop>PeakList. 175 ppm is assigned to all 13C' shifts of SRD-II. | ||
#In XEASY, use <tt>ns</tt> to load both HNNCO spectra <tt>HNCO1</tt> and <tt>HNCO1a</tt> with permutation <tt>x:HN, y:C, Z:N</tt> and <tt>x:N, y:C, Z:HN</tt>, respectively. from <tt>/protName/xeasy/data</tt>; use <tt>ls</tt> to load the sequence file, <tt>nhsqc.seq</tt>; use <tt>lc</tt> to load the atom list (protlist), <tt>nhsqcO1.prot</tt>; use <tt>lp</tt> to load the initial HNCO peaklist from <tt>hncoI1.peak</tt>; use <tt>cp</tt> to display the spectrum as a contour plot; use <tt>se</tt> to create strips for all of the peaks in the peaklist; use <tt>gs</tt> to display the first set of strips (Figure 2A). <br>'''Figure 2: Analysis of the 3D HNNCO by XEASY''' <br>'''A: Before <tt>mr</tt>, showing orthogonal views of each strip;''' <br>[[ | #In XEASY, use <tt>ns</tt> to load both HNNCO spectra <tt>HNCO1</tt> and <tt>HNCO1a</tt> with permutation <tt>x:HN, y:C, Z:N</tt> and <tt>x:N, y:C, Z:HN</tt>, respectively. from <tt>/protName/xeasy/data</tt>; use <tt>ls</tt> to load the sequence file, <tt>nhsqc.seq</tt>; use <tt>lc</tt> to load the atom list (protlist), <tt>nhsqcO1.prot</tt>; use <tt>lp</tt> to load the initial HNCO peaklist from <tt>hncoI1.peak</tt>; use <tt>cp</tt> to display the spectrum as a contour plot; use <tt>se</tt> to create strips for all of the peaks in the peaklist; use <tt>gs</tt> to display the first set of strips (Figure 2A). <br>'''Figure 2: Analysis of the 3D HNNCO by XEASY''' <br>'''A: Before <tt>mr</tt>, showing orthogonal views of each strip;''' <br>[[Image:XEASY hnco1.jpg]] <br> | ||
#In XEASY, use <tt>mr</tt> to move each pre-positioned peak onto the actual peak (Figure 2B); use <tt>dp</tt> to delete unobserved predicted peaks (side-chain peaks), and use <tt>pp</tt> manually pick and assign observed unpredicted peaks (overlapped in Nhsqc).<br>'''B: After <tt>mr</tt>;''' <br>[[ | #In XEASY, use <tt>mr</tt> to move each pre-positioned peak onto the actual peak (Figure 2B); use <tt>dp</tt> to delete unobserved predicted peaks (side-chain peaks), and use <tt>pp</tt> manually pick and assign observed unpredicted peaks (overlapped in Nhsqc).<br>'''B: After <tt>mr</tt>;''' <br>[[Image:XEASY hnco2.jpg]] <br> | ||
#After all peaks are 'picked', use <tt>ac</tt> to update the chemical shifts, <tt>wc</tt> to save the atom list as <tt>hncoO1.prot</tt>, and <tt>wp</tt> to save the peaklist as <tt>hncoO1.peaks</tt>. | #After all peaks are 'picked', use <tt>ac</tt> to update the chemical shifts, <tt>wc</tt> to save the atom list as <tt>hncoO1.prot</tt>, and <tt>wp</tt> to save the peaklist as <tt>hncoO1.peaks</tt>. | ||
#Go to next step for [[XEASY Backbone Assignment|backbone assignment]] | #Go to next step for [[XEASY Backbone Assignment|backbone assignment]] | ||
Revision as of 18:39, 10 November 2009
Spin System Identification
2D [15N,1H]-HSQC provide pairs of correlated amide 15N / 1HN chemical shifts. They seed spin systems - spins of individual residues, whose assignment to the protein sequence is normally not known a priori. In Xeasy-based GFT approach spin systems are called SRD spin systems.
(3,2)D HNNCO or 3D HNNCO provide additional resolution when both 15N and 1HN chemical shifts overlap, and help exclude side-chain peaks. Although 13C' chemical shifts are seldom used for sequence-specific assignment, they are used by CSI and TALOS programs.
Spin System Identification with XEASY/UBNMR
Peak Picking the (15N,1H) HSQC Spectrum
- Go to /protName/analysis/xeasy/nhsqc. Create a file with the amino acids sequence in FASTA format, obtained, for example, from the SPINE web page, and save it as aa.seq. In UBNMR, run makeSeq. This generates an XEASY <nop>SequenceList as nhsqc.seq . This <nop>SequenceList contains entries for the amino acids residues first, followed by two sets of SRDs named in the following SRD-I (Starting from 201) and SRD-II (starting from 401). SRD-II entires serve to handle sequential connectivities. See XEASY Files for Book-keeping: <nop>AtomList, <nop>SequenceList, <nop>PeakList.
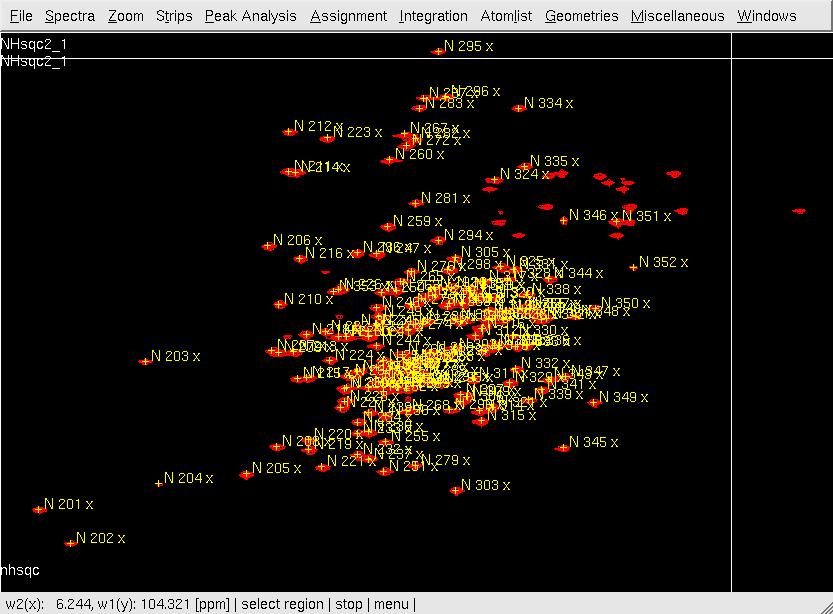
- In XEASY, use ns, ll and ls to load 2D [15N,1H]-HSQC spectrum, XEASY library and <nop>SequenceList. To change display from the default one to contour plot, type cp and give the appropriate threshold level. A corresponding initial <nop>AtomList is generated automatically; use in for in-phase 'peak picking' of the 2D [15N,1H]-HSQC spectrum automatically; complete peak picking manually by using dp to remove the peaks belonging to sidechain amides which can be identified by a NH2only HSQC if the region is crowded and pp to 'peak pick' additional peaks (Figure.1A); Use ar to automatically assign each peak to the backbone amide moiety of SRD-I residues (Figure.1B,C); use ac, wc and wp to save the updated <nop>AtomList as nhsqcO1.prot and <nop>PeakList as nhsqcO1.peaks. Then, 15N / amide 1HN chemical shifts are transferred into the <nop>AtomList entries corresponding to SRD-I. At times the ar command gives an error "Atom N 201 not known!". This can be rectified by loading a different library file [ll] from /nsm/chem/cen2/HTP2/3_src/xeasy/src.new/xeasy.lib and then trying to auto assign the peaks.
Figure 1. Peak picking the NHSQC spectrum*
A: After in-phase peak picking; 
B: XEASY ar window

C: After XEASY command ar .

- In UBNMR run makeHncoPeak. A starting peak list for analysis of (3,2)D HNNCO or 3D HNNCO is generated as hncoI1.peaks. /
- In XEASY, perform HNNCO Analysis (described in HNNCO analysis).
Analysis of the 3D HNNCO Spectrum
- Go to /protName/analysis/xeasy/hnco
- In the spectra folder, make a identical copy of HNCO spectrum, eg. cp spectrum HNCO1 to spectrum HNCO1a.
- In UBNMR, run makeHncoPeak to produce the file hncoI1.peaks. This <nop>PeakList will contain one peak for each amide N,H moiety assigned to SRD-I derived from the 2D [15N,1H]-HSQC <nop>PeakList. 175 ppm is assigned to all 13C' shifts of SRD-II.
- In XEASY, use ns to load both HNNCO spectra HNCO1 and HNCO1a with permutation x:HN, y:C, Z:N and x:N, y:C, Z:HN, respectively. from /protName/xeasy/data; use ls to load the sequence file, nhsqc.seq; use lc to load the atom list (protlist), nhsqcO1.prot; use lp to load the initial HNCO peaklist from hncoI1.peak; use cp to display the spectrum as a contour plot; use se to create strips for all of the peaks in the peaklist; use gs to display the first set of strips (Figure 2A).
Figure 2: Analysis of the 3D HNNCO by XEASY
A: Before mr, showing orthogonal views of each strip;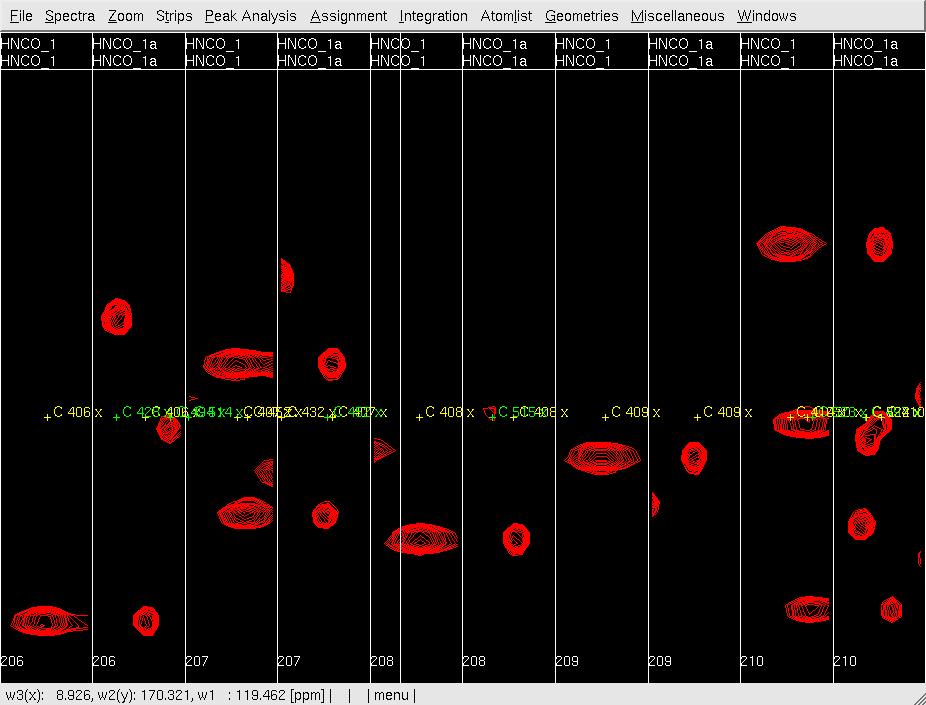
- In XEASY, use mr to move each pre-positioned peak onto the actual peak (Figure 2B); use dp to delete unobserved predicted peaks (side-chain peaks), and use pp manually pick and assign observed unpredicted peaks (overlapped in Nhsqc).
B: After mr;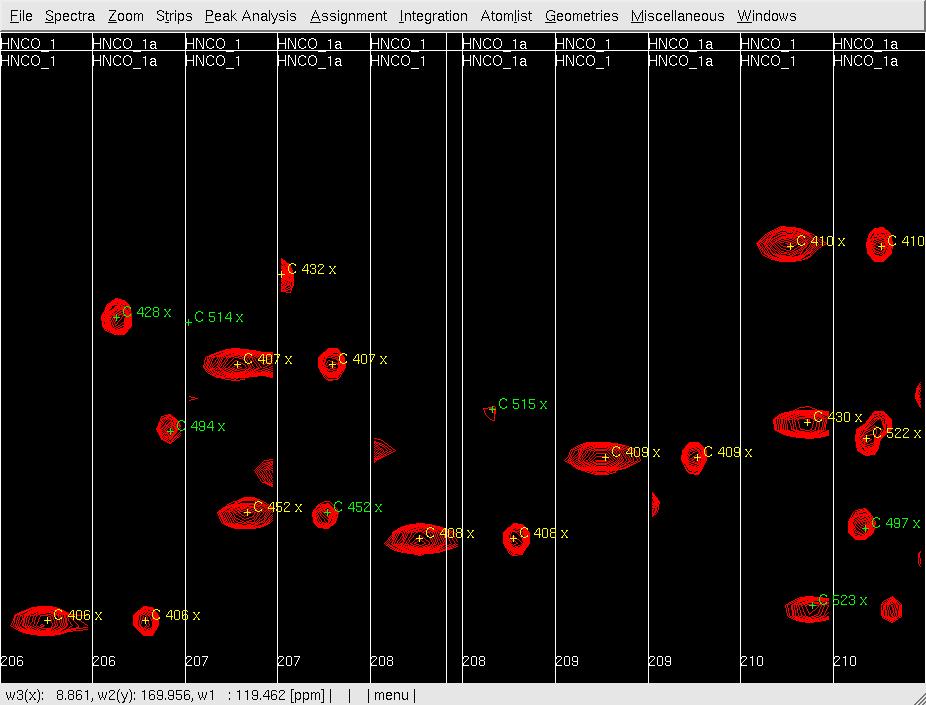
- After all peaks are 'picked', use ac to update the chemical shifts, wc to save the atom list as hncoO1.prot, and wp to save the peaklist as hncoO1.peaks.
- Go to next step for backbone assignment


Analysis of the (3,2)D GFT HNNCO spectrum
- Go to /protName/analysis/xeasy/hnco
- In UBNMR run macro make32DHncoPeak: listed as following to get (3,2)D GFT HNNCO peaklist.
init read seq nhsqc.seq read prot noe.prot write prot hnco.prot simulate 2D N+pC HN simulate 2D N-pC HN write peaks HNNCO.peaks - Do the analysis in XEASY on the (3,2)D GFT HNNCO spectrum similar to that of 3D HNNCO spectrum.
%COMMENT%
-- Main.GaohuaLiu - 16 Feb 2007