XEASY Spin system identification: Difference between revisions
No edit summary |
No edit summary |
||
| Line 13: | Line 13: | ||
==== '''Peak Picking the (15N,1H) HSQC Spectrum''' ==== | ==== '''Peak Picking the (15N,1H) HSQC Spectrum''' ==== | ||
#Go to <tt>/protName/analysis/xeasy/nhsqc.</tt> Create a file with the amino acid sequence in [[ | #Go to <tt>/protName/analysis/xeasy/nhsqc.</tt> Create a file with the amino acid sequence in [[Fasta|FASTA]] format, obtained, for example, from the [http://spine.nesg.org/ SPINE] web page, and save it as <tt>aa.seq</tt>. Run the <tt>makeSeq</tt> macro with [[UBNMR|UBNMR]] to generate an [[XEASY|XEASY]] [[XEASY Sequence List|SequenceList]] as <tt>nhsqc.seq</tt> . This [[XEASY Sequence List|SequenceList]] contains entries for the amino acids residues first, followed by two sets of '''SRD'''s named in the following way: '''SRD-I''' (Starting from 201) and '''SRD-II''' (starting from 401). '''SRD-II''' entires serve to handle sequential connectivities. See [[XEASY|XEASY]] Files for book-keeping: [[XEASY_Atom_List|AtomList]], [[XEASY_Sequence_List|SequenceList]], and [[XEASY_Peak_List|PeakList]]. | ||
#In XEASY, use <tt>ns</tt>, <tt>ll</tt> and <tt>ls</tt> to load 2D [15N,1H]-HSQC spectrum, XEASY library and | #In XEASY, use <tt>ns</tt>, <tt>ll</tt> and <tt>ls</tt> to load 2D [15N,1H]-HSQC spectrum, [[XEASY|XEASY]] library and [[XEASY_Sequence_List|SequenceList]]. To change display from the default one to contour plot, type <tt>cp</tt> and give the appropriate threshold level. A corresponding initial [[XEASY_Atom_List|AtomList]] is generated automatically. Use <tt>in</tt> for automatic in-phase peak picking of the 2D [15N,1H]-HSQC spectrum. Complete peak picking manually by using <tt>dp</tt> to remove the peaks belonging to sidechain amides which can be identified by a NH2only HSQC if the region is crowded and <tt>pp</tt> to pick additional peaks (Figure.1A). Use <tt>ar</tt> to automatically assign each peak to the backbone amide moiety of '''SRD-I''' residues (Figure.1B,C). Occasionally, the <tt>ar</tt> command causes an error <tt>Atom N 201 not known!</tt> which can be solved by loading the library file [<tt>ll</tt>] from <tt>/nsm/chem/cen2/HTP2/3_src/xeasy/src.new/xeasy.lib</tt> and then repeating the <tt>ar</tt> command. Average the chemical shifts and save the [[XEASY_Peak_List|PeakList]] (<tt>nhsqcO1.peaks</tt>) and [[XEASY_Atom_List|AtomList]] (<tt>nhsqcO1.prot</tt>) using <tt>ac</tt>, <tt>wc</tt> and <tt>wp</tt>. Then, amide 15N / 1HN chemical shifts are transferred into the [[XEASY_Atom_List|AtomList]] entries corresponding to '''SRD-I'''. | ||
''' | <br><br>'''Figure 1.''' Peak picking the 2D [15N, 1H]-HSQC spectrum | ||
''' | '''A: After in-phase peak picking'''<br> [[Image:XEASY hsqc2.jpg]] <br> | ||
<br> | |||
<br> [[Image:XEASY hsqc ar.jpg]]<br> | '''B: XEASY <tt>ar</tt> window'''<br> [[Image:XEASY hsqc ar.jpg]]<br> | ||
<br> '''C: After | <br> '''C: After assigning the peaks to SRD-I numbers using <tt>ar</tt>'''<br> | ||
<br> | [[Image:XEASY hsqc3.jpg]]<br> | ||
==== '''Analysis of the 3D HNNCO Spectrum''' ==== | ==== '''Analysis of the 3D HNNCO Spectrum''' ==== | ||
#Run the <tt>makeHncoPeak</tt> script in [[UBNMR|UBNMR]]. A starting peak list for analysis of (3,2)D HNNCO or 3D HNNCO is generated as <tt>hncoI1.peaks</tt>. | |||
#In XEASY, perform HNNCO Analysis (described in HNNCO analysis). | |||
#Go to <tt>/protName/analysis/xeasy/hnco</tt> | #Go to <tt>/protName/analysis/xeasy/hnco</tt> | ||
#In the spectra folder, make a identical copy of HNCO spectrum, eg. cp spectrum <tt>HNCO1</tt> to spectrum <tt>HNCO1a</tt>. | #In the spectra folder, make a identical copy of HNCO spectrum, eg. cp spectrum <tt>HNCO1</tt> to spectrum <tt>HNCO1a</tt>. | ||
Revision as of 18:01, 24 November 2009
Spin System Identification
2D [15N,1H]-HSQC provide pairs of correlated amide 15N / 1HN chemical shifts. They seed spin systems - spins of individual residues, whose assignment to the protein sequence is normally not known a priori. In an XEASY-based approach, spin systems are called SRD spin systems.
(3,2)D HNNCO or 3D HNNCO provide additional resolution when both 15N and 1HN chemical shifts overlap, and help exclude side-chain peaks. Although 13C' chemical shifts are seldom used for sequence-specific assignment, they are used by the programs CSI and TALOS.
Spin System Identification with XEASY/UBNMR
Peak Picking the (15N,1H) HSQC Spectrum
- Go to /protName/analysis/xeasy/nhsqc. Create a file with the amino acid sequence in FASTA format, obtained, for example, from the SPINE web page, and save it as aa.seq. Run the makeSeq macro with UBNMR to generate an XEASY SequenceList as nhsqc.seq . This SequenceList contains entries for the amino acids residues first, followed by two sets of SRDs named in the following way: SRD-I (Starting from 201) and SRD-II (starting from 401). SRD-II entires serve to handle sequential connectivities. See XEASY Files for book-keeping: AtomList, SequenceList, and PeakList.
- In XEASY, use ns, ll and ls to load 2D [15N,1H]-HSQC spectrum, XEASY library and SequenceList. To change display from the default one to contour plot, type cp and give the appropriate threshold level. A corresponding initial AtomList is generated automatically. Use in for automatic in-phase peak picking of the 2D [15N,1H]-HSQC spectrum. Complete peak picking manually by using dp to remove the peaks belonging to sidechain amides which can be identified by a NH2only HSQC if the region is crowded and pp to pick additional peaks (Figure.1A). Use ar to automatically assign each peak to the backbone amide moiety of SRD-I residues (Figure.1B,C). Occasionally, the ar command causes an error Atom N 201 not known! which can be solved by loading the library file [ll] from /nsm/chem/cen2/HTP2/3_src/xeasy/src.new/xeasy.lib and then repeating the ar command. Average the chemical shifts and save the PeakList (nhsqcO1.peaks) and AtomList (nhsqcO1.prot) using ac, wc and wp. Then, amide 15N / 1HN chemical shifts are transferred into the AtomList entries corresponding to SRD-I.
Figure 1. Peak picking the 2D [15N, 1H]-HSQC spectrum
A: After in-phase peak picking

B: XEASY ar window

C: After assigning the peaks to SRD-I numbers using ar

Analysis of the 3D HNNCO Spectrum
- Run the makeHncoPeak script in UBNMR. A starting peak list for analysis of (3,2)D HNNCO or 3D HNNCO is generated as hncoI1.peaks.
- In XEASY, perform HNNCO Analysis (described in HNNCO analysis).
- Go to /protName/analysis/xeasy/hnco
- In the spectra folder, make a identical copy of HNCO spectrum, eg. cp spectrum HNCO1 to spectrum HNCO1a.
- In UBNMR, run makeHncoPeak to produce the file hncoI1.peaks. This <nop>PeakList will contain one peak for each amide N,H moiety assigned to SRD-I derived from the 2D [15N,1H]-HSQC <nop>PeakList. 175 ppm is assigned to all 13C' shifts of SRD-II.
- In XEASY, use ns to load both HNNCO spectra HNCO1 and HNCO1a with permutation x:HN, y:C, Z:N and x:N, y:C, Z:HN, respectively. from /protName/xeasy/data; use ls to load the sequence file, nhsqc.seq; use lc to load the atom list (protlist), nhsqcO1.prot; use lp to load the initial HNCO peaklist from hncoI1.peak; use cp to display the spectrum as a contour plot; use se to create strips for all of the peaks in the peaklist; use gs to display the first set of strips (Figure 2A).
Figure 2: Analysis of the 3D HNNCO by XEASY
A: Before mr, showing orthogonal views of each strip;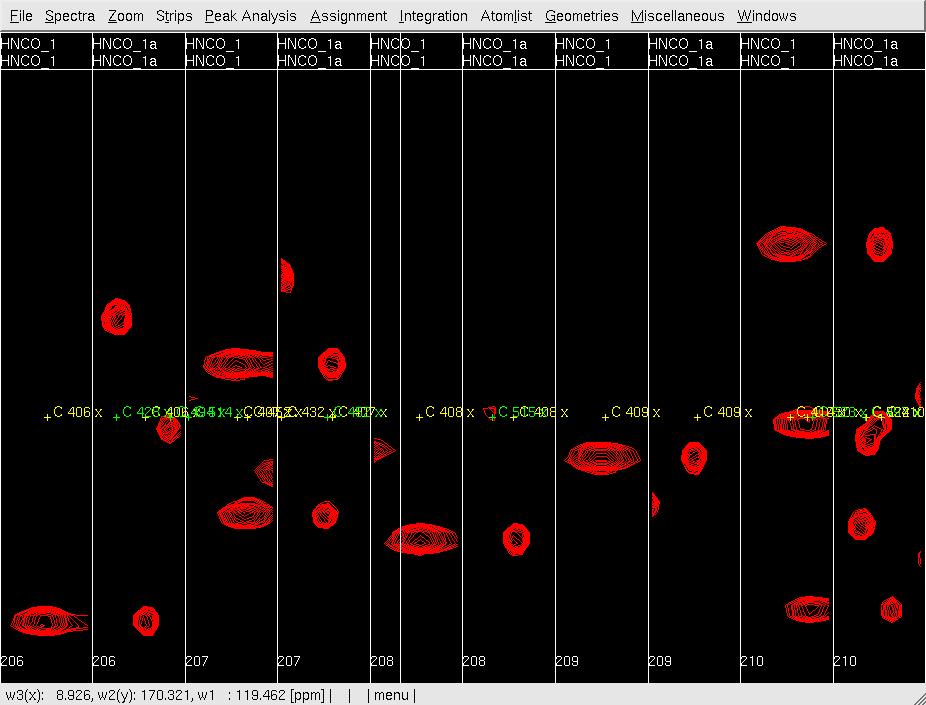
- In XEASY, use mr to move each pre-positioned peak onto the actual peak (Figure 2B); use dp to delete unobserved predicted peaks (side-chain peaks), and use pp manually pick and assign observed unpredicted peaks (overlapped in Nhsqc).
B: After mr;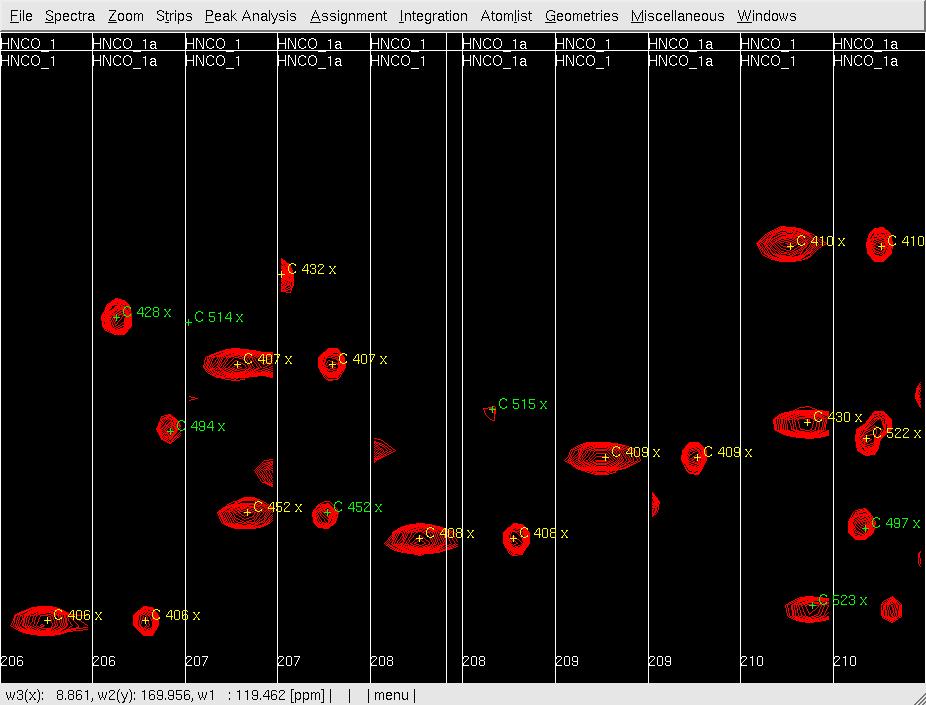
- After all peaks are 'picked', use ac to update the chemical shifts, wc to save the atom list as hncoO1.prot, and wp to save the peaklist as hncoO1.peaks.
- Go to next step for backbone assignment


Analysis of the (3,2)D GFT HNNCO spectrum
- Go to /protName/analysis/xeasy/hnco
- In UBNMR run macro make32DHncoPeak: listed as following to get (3,2)D GFT HNNCO peaklist.
init read seq nhsqc.seq read prot noe.prot write prot hnco.prot simulate 2D N+pC HN simulate 2D N-pC HN write peaks HNNCO.peaks
- Do the analysis in XEASY on the (3,2)D GFT HNNCO spectrum similar to that of 3D HNNCO spectrum.
-- Main.GaohuaLiu - 16 Feb 2007</pre>